
Alkuperäinen teksti (Original Text): Maria Osmala
Blog in English: “What can the genomes of 240 placental mammal species tell us about gene regulation in cancer?”
Nimeni on Maria Osmala, olen bioinformaatikko ja laskennallinen biologi. Olen työskennellyt reilun vuoden professori Jussi Taipaleen Lääketieteellisen systeemibiologian ryhmässä Helsingin yliopiston lääketieteellisessä tiedekunnassa. Minua kiinnostaa erityisesti bioinformatiikka ja laskennalliset menetelmät geenien säätelyn tutkimuksessa. Tässä blogissa pyrin valaisemaan, miten transkriptiotekijöiden sitoutumiskohtien tutkiminen auttaa selvittämään syövän taustalla olevia biologisia mekanismeja. Erityisesti tarkastelen miten transkriptiotekijöiden sitoutumiskohtien säilyneisyys evoluutiossa antaa lisäarvoa tälle tutkimukselle ja miten istukallisten nisäkkäiden syvästi sekvensoituja genomeja voidaan hyödyntää tässä tutkimuksessa.
Geenien ilmentymisen säätely transkriptiotasolla
Jokainen nisäkäslajin solu sisältää genomin eli periytyvän geneettisen tiedon, joka on koodattu deoksiribonukleiinihapon (DNA) nukleotidisekvenssiin. Nukleotidien A, T, C ja G järjestys DNA:ssa sisältää ohjeet esimerkiksi solun kasvulle ja toiminnalle. Vain 2 % genomista muodostuu geeneistä jotka koodaavat proteiineja ja entsyymeitä, jotka suorittavat solujen toiminnallisuuden. Eri geenijoukot ovat aktiivisia eli ilmentyneitä erityyppisissä soluissa, joita ihmiskehossakin on satoja erilaisia, esim. hermosolut ja verisolut. Aktiiviset geenit ja siten kunkin solutyypin erityistoiminnot määrittyvät kuitenkin ei-koodaavien säätelyalueiden kuten promoottoreiden ja varsinkin tehostajien aktiivisuuden mukaan. Tehostajat sijaitsevat hajallaan vähemmän tunnetussa 98 % osuudessa ihmisen genomia, ja niiden sijanti ja rooli eri solutyypeissä ja sairauksien, kuten syövän synnyssä, on suurelta osin kartoittamatta.
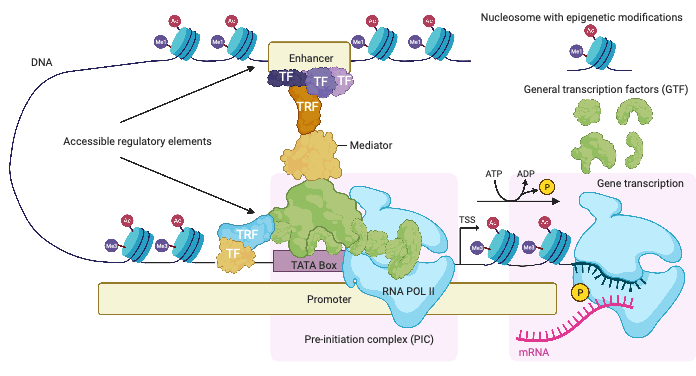
Säätelyalueiden nukleotidijärjestyksen tulkintaan erikoistuneita proteiineja kutsutaan transkriptiotekijöiksi. Transkriptiotekjijät tunnistavat lyhyitä DNA sekvenssejä säätelyalueilla ja sitoutuvat niihin. Lääketieteellisen Systeemibiologian ryhmässä on tutkittu erityisesti transkriptiotekijää nimeltä MYC. MYC sitoutuu lukuisille säätelyalueille ja säätelee suurta määrää eri geenejä, jotka suorittavat ja säätelevät eri toimintoja normaalissa solun kasvussa ja jakautumisessa. Kasvainten muodostumisen aikana nämä säätelymekanismit häiriintyvät ja solut alkavat kasvaa hallitsemattomasti. Suurin osan ihmisen eri syöpätyyppien (> 70%) synnystä on seurausta transkriptiotekijä MYCin virheellisestä ilmentymistasosta. MYC on siten niin sanottu onkogeeni.
MYC geenin ilmentymistä soluissa säätelevät sen promoottorin lisäksi useat tehostaja-alueet. MYCin proteiinirakenne on sellainen, ettei siihen kohdennettua lääkettä ole helppo kehittää. Näiden syiden vuoksi on tärkeää tutkia transkriptiotekijöiden sitoutumiskohtia MYCin säätelyalueella ja MYCin kohdegeenien säätelyalueilla. Tavoitteena on löytää MYCin toiminnalle tärkeitä transkriptiotekijöiden sitoutumiskohtia, tulkita syöpään liittyviä mutaatioita ja riskivariantteja näillä alueilla, ja löytää potentiaalisia lääkekohteita.
Transkriptiotekijät tulkitsevat nukleotidijärjestystä säätelyalueilla ja vuorovaikuttavat toistensa kanssa
Yhteen säätelyalueeseen voi sitoutua useita eri säätelytekijöitä, ja usein ne eivät sitoudu yksinään vaan vuorovaikutuksessa toistensa kanssa. Säätelytekijöiden sitoutumiskohtien jakautuminen säätelyelementillä sekä transkriptiotekijöiden välisten vuorovaikutuksien vaikutus geenien säätelyyn ovat aktiivisen tutkimuksen kohteita. Transkriptiotekijät ovat suurin proteiiniperhe ihmisen soluissa ja erilaisten proteiinien määrä on arviolta noin 2000; täten jo parittaisia yhteisvuorovaikutuksia voi olla miljoonia (2000 * 1999 4 miljoonaa).
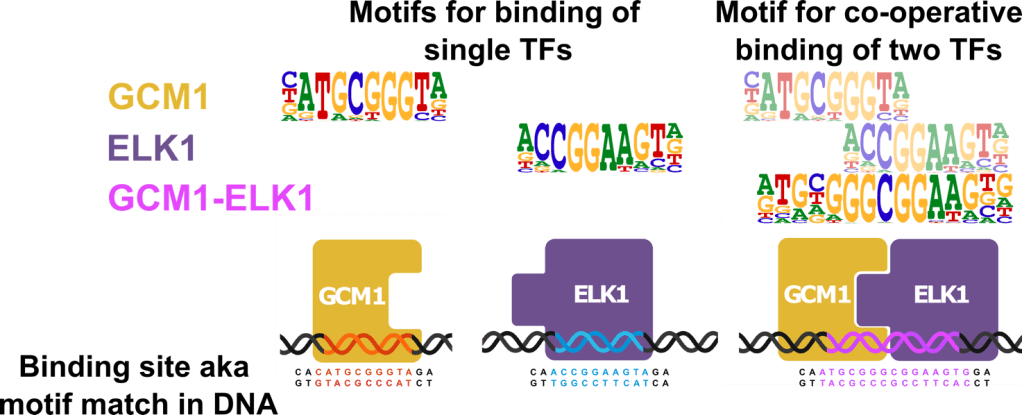
On tärkeää selvittää yksittäisten transkriptiotekijöiden spesifisyys tiettyjä DNA sekvenssejä kohtaan eli motiivit. Erilaisilla määrityksillä voidaan selvittää erityiset tai spesifit DNA sekvenssit, joihin kukin yksittäinen transkriptiotekijä sitoutuu. Transkriptiotekijän sekvenssi-spesifisyys esitetään usein motiivilogona, kuvassa alla esimerkkinä motiivit transkriptiotekijöille GCM1 ja ELK1. Koska transkriptiotekijät usein vuorovaikuttavat keskenään saavuttaakseen sitoutumisen, on tärkeää määrittää myös motiivit kahden transkriptiotekijän yhteistoiminnalliselle sitoutumiselle. Yhteistoiminnalliset motiivit ovat suurelta osin tuntemattomia, ja ne saattavat poiketa vastaavista yksittäisten transkriptiotekijöiden motiiveista, esimerkkinä yhteistoimintamotiivi GCM1-ELK1 parille.
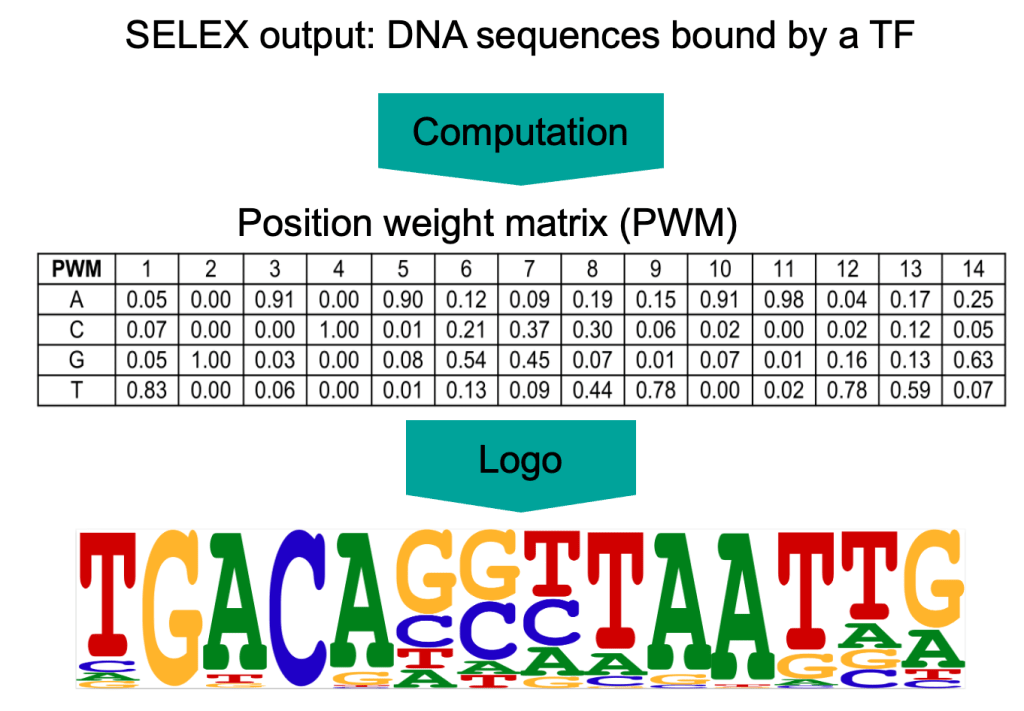
TF-motiivit johdetaan lyhyistä DNA-sekvensseistä, joiden tiedetään sitoutuvan TF:ään. Tällaiset sekvenssit voidaan paljastaa käyttäen määritystä nimeltä Ligandien valikoiva evoluutio eksponentiaalisen rikastumisen avulla (SELEX). Lääketieteellisen systeemibiologian ryhmällä on pitkät perinteet motiivien selvittämiseksi käyttäen SELEX-tekniikkaan perustuvia määrityksiä. SELEX-kokeiden tuloksena saadaan kokoelma lyhyitä DNA sekvenssejä, joihin transkriptiotekijä sitoutuu. Sekvenssien perusteella voidaan tilastollisesti määrittää transriptiofaktorin sekvenssispecifisyys, jota kutsutaan asemapainomatriisiksi (position weight matrix, PWM) tai motiiviksi. Asemapainomatriisi kertoo todennäköisyyden kullekin neljälle emäkselle esiintyä jokaisessa sitoutumiskohdan asemassa.
Lääketieteellisen systeemibiologian ryhmän tuottama SELEX-motiivien kokoelma sisältää noin tuhat erilaista transkriptiofactorien sekvenssispesifisyyttä. Kukin motiivi voidaan skannata koko ihmisen genomin läpi paikallistaaksemme kaikki mahdolliset yksittäisten tai yhteistyössä toimivien transkriptiotekijöiden sitoutumiskohdat. Mahdollisia sitoutumiskohtia kutsutaan myös motiiviosumiksi. Koska transkriptiotekijöiden sitoutuminen DNA:han riippuu useista tekijöistä (katso kuva 1), vain osa motiiviosumista on toiminnallisia riippuen solun tyypistä ja tilasta. Lisänäyttöä motiiviosumien toiminnallisuudesta saadaan hyödyntämällä vertailevaa genomiikka lajien välillä ja DNA:n säilyneisyyttä evoluutiossa.
Vertaileva genomiikka: Säätelyalueiden DNA:n säilyminen istukkanisäkkäiden evoluutiossa
Yksi tapa tulkita ei-koodaavaa DNA:ta on tarkastella ihmisen genomin alkuperää evoluutioteorian kautta. Teorian mukaan kaikki istukkanisäkkäät mukaan lukien ihminen ovat sukua toisilleen. Muutokset eli mutaatiot genomeissa lajien välillä johtuvat epätäydellisestä perimän kopioinnista sukusolulinjassa ja luonnonvalinnasta. Mutaatiot voivat olla hyödyllisiä, haitallisia tai neutraaleja, ja ne voivat johtaa muutoksiin organismin havaittavissa olevissa ominaisuuksissa. Nukleotidit, jotka ovat säilyneet samoina evoluution ajan, ovat todennäköisesti toiminnallisesti tärkeitä. Näillä säilyneillä genomialueilla esiintyy hitaampaa evoluutiota kuin voisi tilastollisesti odottaa. Säilyneiden genomialueiden etsiminen, jota kutsutaan myös vertailevaksi genomiikaksi, on tehokas tapa tunnistaa säätelyelementit genomista ja kartoittaa toiminnallisia muutoksia DNA:ssa. Säilyneillä alueilla ihmisen genomissa esiintyy sekä sukusolulinjassa että somaattisissa soluissa kartoitettuja DNA muunnelmia ja mutaatioita, jotka on liitetty monimutkaisten sairauksien syntyyn. Vertaileva genomiikka voi siten edistää biolääketiedettä.

Suurin resurssi istukkaeläinten vertailevaan genomianalyysiin on äskettäin valmistunut Zoonomia-projekti, joka syväsekvensoi 240 lajin genomit ja suoritti sekvenssien laskennallisen analyysin. Laskennallinen analyysi sisältää tilastollisen evoluutiomallin sovittamisen genomiaineistoon. Evoluutiomallin taustalla on paljon oletuksia ja yksinkertaistuksia, mutta malli on todettu hyödylliseksi. Saadun mallin perusteella voidaan testata tilastollisesti esim. minkä tahansa ihmisgenomin nukleotidin kohdalla, onko nukleotidi säilynyt evoluutiossa tietyllä merkitysevyystasolla. Tuloksena saatua p-arvoa logaritmisella asteikolla kutsutaan PhyloP pistearvoksi.
Työssäni tutkin miltä nämä PhyloP pistearvot näyttävät SELEX motiivien osumilla eli mahdollisilla transkriptiotekijöiden sitoutumiskohdilla ihmisen genomissa. Korkeat genomi-osumien PhyloP pistearvot tuovat lisätodistetta siihen, että motiivit ja niiden genomi-osumat todella ovat toiminnallisia soluissa. On myös mielenkiintoista tutkia esiintyykö konservoituneiden motiivi-osumien tietyissä sitoutumiskohdan asemissa korkeampia PhyloP pisterarvoja kuin toisissa; nämä asemat saattavat olla transkriptiotekijän ja DNA:n vuorovaikutuksessa erityisen tärkeitä ja mutaatiot näissä häiritsevät tekijöiden sitoutumista ja säätelyalueen toimintaa.
Eteenpäin suuntautuva genomiikka (Forward genomics): miksi jotkin istukalliset nisäkkäät ovat resistenttejä syövälle?
Toinen tapa, millä istukallisten nisäkkäiden genomien tutkimus voi edistää biolääketiedettä on ns. ”Forward genomics” lähestymistapa. Forward genomics tutkimuksessa tunnistetaan nisäkäsryhmässä lääketieteellisesti merkityksellinen ominaisuus. Vertailemalla genomeja kahden nisäkäsryhmän välillä, joissa toisessa ominaisuus esiintyy ja toisessa ei, voidaan tunnistaa DNA-alueet, jotka todennäköisesti aiheuttavat ominaisuuden. Eräs mielenkiintoinen ominaisuus on tiettyjen nisäkkäiden vastustuskyky syövälle, vaikka ne ovat suurikokoisia ja elävät pitkään. Suurikokoisen ja pitkään elävän nisäkkään solut käyvät läpi enemmän solunjakautumista, ja täten voisi olettaa että näiden soluilla on suurempi mahdollisuus kerryttää haitallisia mutaatioita, jotka aiheuttavat syöpää. Kuitenkaan suurilla nisäkäslajeilla syöpäriski ei ole suurempi ja jotkin lajit pystyvät vastustamaan syöpää hyvin tehokkaasti. Tämä ilmiö tunnetaan nimellä Peton paradoksi, Brittiläisen epidemiologin Richard Peton mukaan. Tutkijat ovat havainneet, että esimerkiksi sorkkaeläimet, erityisesti märehtijät, norsut, valaat ja suurin jyrsijä kapybara eli vesisika ovat erityisen vastustuskykyisiä syövän synnylle.

Norsut elävät lähes yhtä pitkään kuin ihmiset, mutta kuolevat harvoin syöpään. Tutkijat havaitsivat, että norsujen soluissa on ylimääräisiä kopioita tuumorisuppressorigeenistä nimeltä TP53. Yhdessä muiden löydettyjen geenien kanssa TP53 saattaa estää DNA-vaurioiden vaikutuksia tuhoamalla vaurioituneita soluja. Yksi suurimmista, painavimmista ja pisimpään elävistä nisäkkäistä on keulapäävalas, jonka arvellaan elävän yli 200 vuotiaaksi. Keulapäävalas vastustaa syöpää myös tehokkaasti. Tämän arvellaan johtuvat tarkemmasta ja tehokkaammasta DNA-vaurioiden korjausmekanismista. Eri lajien elinikää ja riskiä sairastua ja kuolla syöpään on ollut haastavaa tutkia, mutta nyt tutkijoiden käytössä on Species360 organisaation keräämä hyödyllinen tietokanta eläintarhoissa elävien nisäkkäiden syöpätapauksista ja muista terveystiedoista.
Tavoitteeni on tutkia evoluutiossa säilyneiden alueiden ja forward genomics tutkimuksessa määritettyjen alueiden päällekkäisyyttä MYC onkogeenin sekä sen kohdegeenien säätelyelementtien kanssa. Signaalit evolutiivisesta säilymisestä näillä alueilla auttavat transkriptiotekijöiden sitoutumiskohtien priorisoinnissa. Lisäksi on mielenkiintoista tutkia vastaavia MYC:in ja kohdegeenien säätelyalueita niiden nisäkkäiden genomeissa, jotka ovat poikkeuksellisen resistenttejä syövälle; tämä voisi valaista myös näiden alueiden toimintaa ihmisgenomissa ja auttaa syöpään liittyvien mutaatioiden tulkinnassa ja edelleen kohdennettujen lääkkeiden kehittämisessä. Lääketieteellisen systeemibiologian ryhmä tutkii myös laajasti MYC:in ja sen kohdegeenien säätelyalueiden toimintaa paksusuolen syövässä käyttäen syöpäsolu- ja hiirimalleja. Laskennallisen analyysin löydöksiä voidaan kokeellisesti vahvistaa näissä malleissa.
Voit seurata meitä: Twitter (@CoEinTG) ja Youtube

One thought on “Mitä 240 istukkanisäkäslajin genomit voivat kertoa meille geenien säätelystä syövässä?”
Comments are closed.