
By: Maria Osmala
Suomennos: Mitä 240 istukkanisäkäslajin genomit voivat kertoa meille geenien säätelystä syövässä?
My name is Maria Osmala, I’m a bioinformatician and a computational biologist in Professor Jussi Taipale’s Medical Systems Biology group at the University of Helsinki. I am interested in bioinformatics and computational research on the regulation of gene expression. Here, I aim to shed light on how research on transcription factor binding sites helps to unravel the biological mechanisms underlying cancer. Specifically, I examine how the evolutionary conservation of transcription factor binding sites adds value to this research, and how the deeply sequenced genomes of placental mammals can be utilized in this study.
Gene expression regulation at the transcription level
Each mammalian cell contains the genome, the heritable genetic information, encoded in the nucleotide sequence of deoxyribonucleic acid (DNA). The specific order, or sequence, of the nucleotides A, T, C, and G contains the instructions e.g., for the growth and function of the cell. Only 2% of the genome comprises of genes that code for proteins and enzymes that carry out cellular functions. Different sets of genes are active, i.e., expressed in each cell type, of which there are hundreds in the human body, such as neurons and blood cells. However, the set of active genes, and thus the specific functions of each cell type, are determined by the activity of non-coding regulatory regions such as promoters and especially enhancers. Enhancers are scattered over a less known 98% of the human genome, and their location and role in different cell types and in the development of diseases such as cancer is largely uncharted.
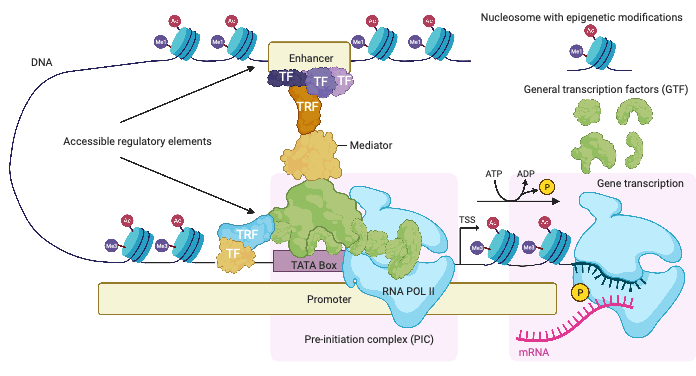
Proteins specialized in interpreting the nucleotide sequence at regulatory elements are called transcription factors (TFs). TFs recognize short DNA sequences at regulatory elements and bind to them. The Medical Systems Biology group specifically studies a TF called MYC. MYC binds to numerous regulatory elements and regulates many different genes, which perform and regulate various functions in normal cell growth and division. During tumor formation, these regulatory mechanisms are disrupted, and cells begin to grow uncontrollably. Most of the onset of different types of human cancer (> 70%) is the result of incorrect expression levels of MYC. MYC is thus an oncogene.
The expression of the MYC gene in cells is regulated by its promoter and several enhancer elements. The protein structure of MYC is such that it is not easy to develop a drug that targets it. For these reasons, it is important to study the binding sites of TFs at regulatory elements of MYC itself and its target genes. The goal is to find important TF binding sites for the function of MYC, to interpret mutations and risk variants associated with cancer at these elements, and to identify potential drug targets.
Transcription factors interpret the nucleotide sequence at regulatory elements and co-operate with each other
Several different TFs can bind to a single regulatory element, and often they do not bind alone but co-operate in binding. The distribution of TF binding sites on the regulatory element and the impact of TF interactions on gene regulation are areas of active research. TFs are the largest family of proteins in human cells, and the number of different proteins is estimated to be around 2000; thus, even pairwise interactions can number in the millions (2000 * 1999 ≈ 4 million).
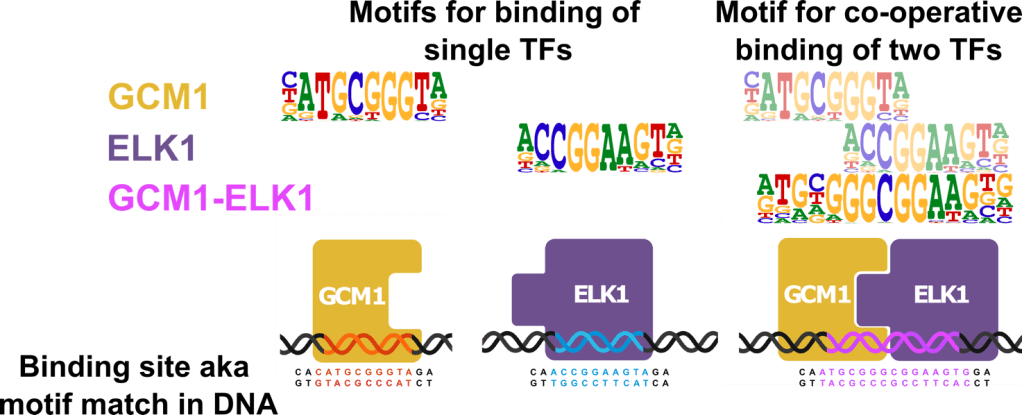
It is important to determine the specificity of TFs towards certain DNA sequences, also known as motifs. Various assays can be used to identify the specific DNA sequences to which each individual TF binds. The sequence specificity of a TF is often represented as a motif logo, the example below shows motifs for TFs GCM1 and ELK1. Since TFs often interact with each other to achieve binding, it is also important to determine motifs for the co-operative binding of two TFs. The motifs for interaction are largely unknown and may differ from the corresponding motifs of individual transcription factors, the example shows the motif for GCM1-ELK1 interaction pair.
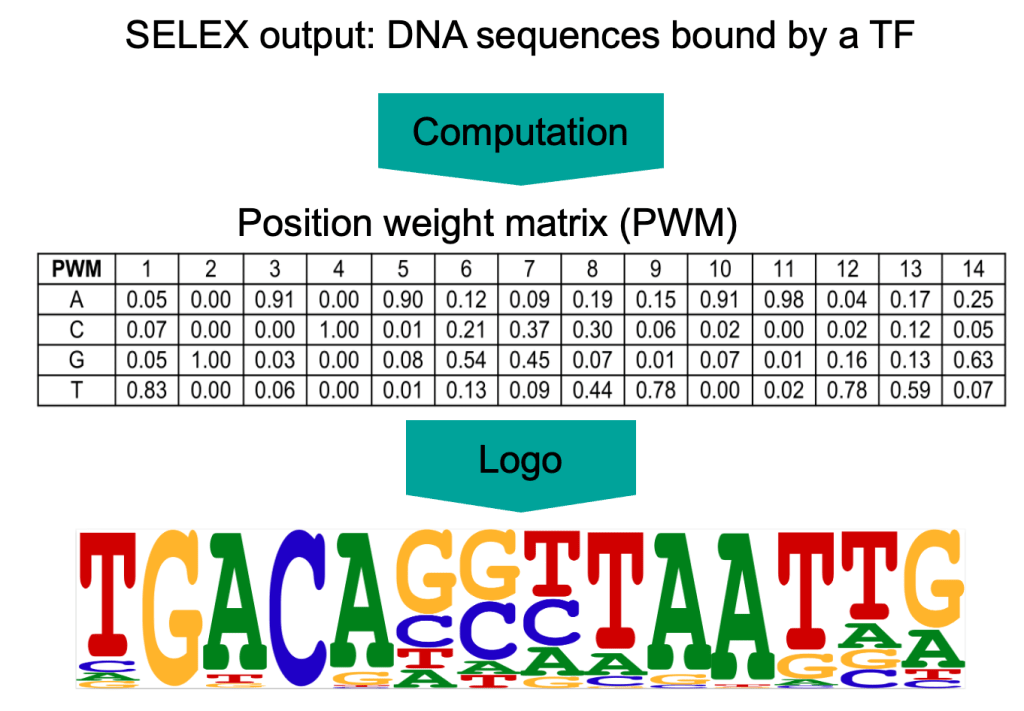
TF motifs are derived from short DNA sequences known to bind to TF. Such sequences can be revealed by high-throughput systematic evolution of ligands by exponential enrichment assay (SELEX). The Medical Systems Biology group has a long tradition of elucidating motifs using experiments based on the SELEX technique. SELEX experiments result in a collection of short DNA sequences to which a TF binds. From these sequences, the sequence specificity of the TF can be statistically determined, called the positional weight matrix (PWM) or motif. The PWM indicates the probability of each of the four bases occurring at each position of the binding site.
The SELEX motif collection produced by the Medical Systems Biology group contains about a thousand different TF sequence specificities. Each motif can be scanned through the entire human genome to locate all possible binding sites for individual or co-operating TFs. This is also termed as motif matching and the identified binding sites as motif matches or hits. Since the binding of TFs to DNA depends on several factors (see Figure 1), only a subset of motif matches is functional depending on the cell type and state. Additional evidence of the functionality of motif matches is obtained by utilizing comparative genomics between species and the conservation of DNA in evolution.
Comparative genomics: conservation of regulatory DNA in the evolution of placental mammals
One way to interpret non-coding DNA is to look at the origin of the human genome through evolutionary theory. According to this theory, all placental mammals are related to each other, and changes, or mutations, in genomes between species are due to imperfect replication of the genome in the germ line and natural selection. Mutations can be beneficial, harmful or neutral, and can lead to changes in observable characteristics of the organism, also known as a trait. Nucleotides that have remained the same over evolutionary history are likely to be functionally important. These conserved genomic regions show slower evolution than would be statistically expected. The search for conserved genomic regions, also called comparative genomics, is an effective way to identify regulatory elements in the genome and map functional changes in DNA. Conserved regions in the human genome contain DNA variations and mutations identified both in germ line and somatic cells that are associated to the development of complex diseases. Comparative genomics can thus contribute to biomedicine.

The largest resource for comparative genomic analysis of placental mammals is the recently completed Zoonomia project, which sequenced deeply the genomes of 240 species and performed computational analysis of the sequences. This computational analysis involves fitting a statistical evolutionary model to the genomic data. The evolutionary model is based on many assumptions and simplifications but has proven to be useful. Based on the obtained model, it is possible to test statistically, for example, whether any nucleotide in the human genome has been conserved in evolution at a certain level of significance. The resulting logarithmic p-value is called a PhyloP score.
In my work, I study the PhyloP scores at the SELEX-motif matches, the potential TF binding sites in the human genome. High PhyloP scores on motif matches provide additional evidence that the motifs are valid, and the motifs matches are indeed functional in cells. It is also interesting to investigate whether certain positions of the conserved motif matches show higher PhyloP scores than the other; these positions may be particularly important for the interaction between the TF and DNA; mutations in the highly conserved nucleotides may disrupt the binding of the TF and the function of the regulatory element.
Forward genomics: why are some placental mammals resistive to cancer?
Another way in which the study of placental mammal genomes can advance biomedicine is through the so-called “forward genomics” approach. In forward genomics research, a medically relevant trait is identified in a group of mammals. By comparing genomes between two groups of mammals, one of which the trait occurs and in the other does not, genomic locations that likely cause the trait can be identified. One interesting trait is the resistance to cancer of certain mammals, even though they are large and live long. The cells of a large and long-lived mammal undergo more cell divisions, and thus one might expect that these cells have a higher chance of accumulating harmful mutations that cause cancer. However, the risk of cancer is not higher in large mammalian species, and some species are very effective at resisting cancer. This phenomenon is known as Peto’s paradox, named after British epidemiologist Richard Peto. Researchers have observed that, for example, ungulates, especially ruminants, elephants, whales, and the largest rodent, the capybara, are particularly resistant to the development of cancer.

Elephants live almost as long as humans, but rarely die from cancer. Scientists discovered that elephant cells have extra copies of a tumor suppressor gene called TP53. Together with other genes, TP53 may counteract the effects of DNA damage by destroying damaged cells. One of the largest, heaviest and longest-living mammals is the bowhead whale, estimated to live over 200 years. The bowhead whale also effectively resists cancer. This may be due to a more precise and effective DNA damage repair mechanism. The lifespan and risk of disease and death from cancer in different species have been challenging to study, but now researchers have access to a useful database collected by the Species360 organization, which includes cases of cancer and other health information about mammals living in zoos.
My aim is to examine the overlap between evolutionary conserved regions and areas identified through forward genomics research with the regulatory elements of the MYC oncogene and its target genes. Signals of evolutionary conservation in these areas assist in prioritizing transcription factor binding sites. In addition, it is intriguing to examine corresponding regulatory elements of MYC and its target genes in the genomes of mammals that are exceptionally resistant to cancer; this could illuminate the function of these areas in the human genome and help interpret cancer-related mutations and further the development of targeted drugs. The Medical Systems Biology group also extensively studies the regulatory elements of MYC and its target genes in colorectal cancer using cancer cell lines and mouse models. The findings of my computational analysis can be experimentally confirmed in these models.
Want to learn more about the Finnish Center of Excellence in Tumor Genetics?
Keep up to date with the latest tweets, videos and blog posts for a peek into the everyday
life in cancer research; subscribe to our blog and YouTube channel! and don’t forget to follow us on Twitter (@CoEinTG)

One thought on “What Can The Genomes of 240 Placental Mammal Species Tell Us About Gene Regulation in Cancer?”
Comments are closed.